It has recently been found that some RNA functions are determined by the actual folding process itself and not just the RNA’s nucleotide sequence or its native structure. We present new computational tools that can be used to study kinetics-based functions for RNA such as population kinetics, folding rates, and the folding of particular subsequences. Previously, these properties could only be studied for small RNA whose conformation space could be enumerated (e.g., RNA with 30-40 nucleotides) or for RNA whose kinetics were restricted in some way.
RNA folding pathway from misfolded to native that must overcome an energy barrier.
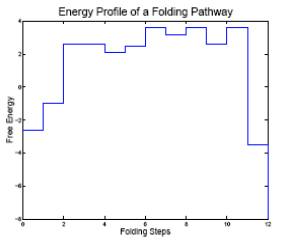
Energy profile for folding pathway.
Our method first builds an approximate map (or model) of the RNA’s folding energy landscape. Then, to study the global folding kinetics, we use the master equation, a matrix of differential equations to describe the population kinetics of the RNA. It allows us to discover features such as folding rates, transitions rates, and folding pathways. Next, to study some low-level folding details, we use our new analysis technique, called Map-based Monte Carlo (MMC) simulation, to stochastically extract folding pathways from the map. MMC, in conjunction with a novel sampling strategy (Boltzmann Statistical Sampling -BSS) for building the map, enables us to study kinetics-based functions for large RNA, e.g., RNA with 200+ nucleotides.
We provide several different simulation results to validate our method against known experimental data. We also show how our method can study kinetics-based functions for two different case studies. First, we compare simulated folding rates for ColE1 RNAII (200 nucleotides) and its mutants against experimental rates and show that our method identifies the same relative folding order as seen experiment. Second, we predict the gene expression rates fo wild-type MS2 phage RNA (135 nucleotides) and three of its mutants and show that our approach computes the same relative functional rates as seen in experiment.
Population Kinetics
To validate our description of the energy landscape using roadmaps, we calculate the population kinetics from our roadmaps and compare them with the results calculated by standard Monte Carlo simulation. Here we show results for the native state of 1k2g (CAGACUUCGGUCGCAGAGAUAGG) using standard Monte Carlo simulation (implemented by Kinfold), Map-based Monte Carlo (MMC) simulation on a fully enumerated Base-pair (BP) roadmap (12.137 conformations), Map-based Monte Carlo (MMC) simulation on a roadmap with our new Boltsmann statistical sampling (BSS) method (42 conformations), and the master equation (ME) on a BSS roadmap (42 conformations). The BP roadmaps is the most accurate representation while the BSS roadmaps yields much smaller subsets of the entire conformation space that effectively approximates the energy landscape.
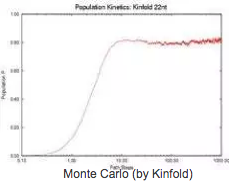



All population kinetics curves have similar features. Also note that the equilibrium (final) distributions are all the same, even though the Boltzmann statistical-sampling roadmap contains less than 0.4% of all possible conformations. Thus, these roadmaps capture the main features of the energy landscape. This data validates that these analysis methods are interchangeable. Quantitative comparisons on master equation results are available in the publications.
Prediction of the Plasmid Replication Rate of ColE1 RNAII
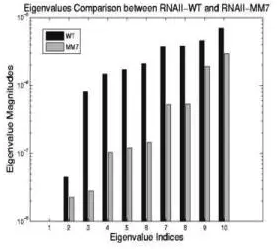
ColE1 RNAII (200 nucleotides) regulates the replication of E. coli ColE1 plasmids through this folding kinetics. The slower it folds, the higher the plasmid replication rate. A specific mutant, MM7, differs from the wild-type (WT) by a single nucleotide out of the 200 nucleotide sequence. This mutation causes it to fold slower while maintaining the same thermodynamics of the native state. Thus, the overall plasmid replication rate increases in the presence of MM7 over the WT.
We compare simulated folding rates for ColE1 RNAII and its mutants against experimental rates. We show the eigenvalues calculated using the master equation. Note that the smallest non-zero eigenvalues correspond to the folding rate. All eigenvalues of WT are larger than MM7 indicating that WT folds faster than MM7. This shows that our method identifies the same relative folding order as seen in experiment.
Prediction of the Gene Expression Rates of MS2 phage RNA
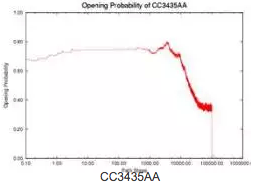
MS2 phage RNA (135 nucleotides) regulates the expression rate of phage MS2 maturation protein at the translational level. It works as a regulator only when a specific subsequence (the SD sequence) is open (i.e., does not form base-pair contacts). Three mutants have been studied that have similar thermodynamic properties with the wild-type (WT) but different kinetics and therefore different gene expression rate. Intuitively, the functional rate (e.g., gene expression rate in this case) is correlated with the opening of the SD sequence. If the SD sequence is opened longer, or has higher opening probability (i.e., having more nucleotides on the SD sequence open), then the mutant should have higher functional rate.
We use our simulation method to study this opening probability during the folding process. Note that mutant CC3435AA has the longest duration at a relatively high level of opening probability while mutant SA has the shortest duration. This correlates with experimental data. The opening probability of U32C decreases earlier but finishes later than WT, so it is not clear which one has a larger total opening probability during folding, again matching experimental findings.



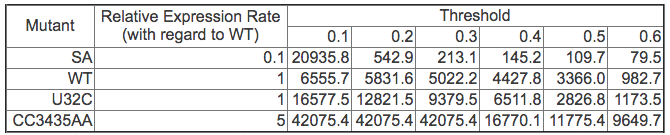
The gene expression rate is determined from two factors: (1) how high the opening probability is at any given time and (2) how long the RNA stays in the high opening probability state. To compare each RNA quantitatively, we compute the integration of the opening probability over the whole folding process. Note that the RNA regulates gene expression only when the SD opening probability is “high enough”. We used thresholds ranging from 0.1 to 0.6 to estimate the gene expression rate. Thresholds higher than 0.6 will yield zero opening probability on WT and most mutants and thus cannot be correlated to experimental results.
The table below shows the results for the WT and for each mutant. For most thresholds, mutant CC3435AA has the highest rate and mutant SA has the lowest rate, the same relative functional rate as seen in experiment. In addition WT and mutant U32C have similar levels (particularly between 0.4-0.6), again correlating with experimental results. Aside from simply validating our method against experiment, we can also use our method to suggest that the SD sequence may only be active for gene regulation when more than 40% of its nucleotides are open.
Comparison of integration of SD opening probability between WT and three mutants of MS2.